Cicloadición 1,3-dipolar
La cicloadición 1,3-dipolar es una reacción de cicloadición en química orgánica. En esta reacción participa una molécula con un fragmento 1,3-dipolar (es decir, un fragmento que forme un dipolo eléctrico a nivel molecular por deslocalización electrónica a lo largo de 3 átomos).[1] Como ejemplos de 1,3-dipolos están el ozono, las azidas, los diazoalcanos, los nitrilos, las nitronas, etc. que se unen a alquenos o alquinos (que serían los dipolarófilos) por cicloadición 1,3-dipolar.[2] La economía atómica de las reacciones de cicloadición 1,3-dipolar es excelente. La cicloadición 1,3-dipolar se esquematiza a continuación:

Actualmente, la cicloadición 1,3-dipolar es una ruta importante para la síntesis regio- y estereoselectiva de heterociclos de cinco miembros y sus derivados acíclicos de anillo abierto.
Rolf Huisgen fue el primero en realizar una investigación sobre esta reacción química mediante la variación del 1,3-dipolo y considerar su importancia en la síntesis de heterocíclicos de cinco eslabones (átomos).[3]

Mecanismo de la reacción editar
Originalmente había dos propuestas que describen el mecanismo de la cicloadición 1,3-dipolar: en primer lugar, el mecanismo de cicloadición pericíclica concertada, propuesto por Rolf Huisgen;[4] y en segundo lugar, un mecanismo por etapas con un intermedio biradical, propuesto por Firestone[5] Después de mucho debate, la propuesta que es ahora generalmente aceptada[6] consiste en una reacción concertada entre el 1,3-dipolo y el dipolarófilo, a menudo de forma asíncrona, y la simetría permitida es π4s + π2s térmica de 6 electrones a través de un estado de transición aromático. Aunque también existen muchos ejemplos de reacciones de cicloadición 1,3-dipolar por pasos.[7][8]
El mecanismo general se describe en la siguiente imagen de forma esquemática:

Consecuencias del mecanismo pericíclico editar
Huisgen investigó una serie de cicloadiciones entre diazocompuestos (1,3-dipolos) y diversos alquenos dipolarófilos.[4] Las siguientes observaciones apoyan el mecanismo pericíclico concertado, y refutan la biradical por etapas.
- Efectos de los sustituyentes: diferentes sustituyentes sobre el dipolo no exhiben un gran efecto sobre la velocidad de cicloadición, lo que sugiere que la reacción no implica un producto intermedio con carga separada.
- Efecto del disolvente: la polaridad del disolvente tiene poco efecto sobre la velocidad de cicloadición, de acuerdo con el mecanismo pericíclico donde la polaridad no cambia mucho al pasar de los reactivos al estado de transición.
- Estereoquímica: las cicloadiciones 1,3-dipolares son siempre estereoespecíficas con respecto al dipolarófilo (es decir, cis-alquenos dan productos-syn), apoyando el mecanismo pericíclico concertado en el que se forman dos enlaces sigma simultáneamente.
- Parámetros termodinámicos: las cicloadiciones 1,3-dipolares tienen una inusual gran entropía negativa en la etapa de activación, de manera similar a la que se da en la reacción de Diels-Alder, lo que sugiere que el estado de transición está altamente ordenado, que es una firma de reacciones pericíclicas concertadas.
Dipolos-1,3 típicos editar

Ya se ha explicado anteriormente que es un 1,3-dipolo (una molécula que forma un dipolo eléctrico a nivel molecular por deslocalización electrónica de 4 electrones en un sistema π a lo largo de 3 átomos).[9]
Se pueden escribir distintas estructuras de resonancia en las que se deslocalizan ambas cargas negativas y positivas en cualquier extremo de un 1,3-dipolo (véase el esquema siguiente). Un método más preciso para describir la distribución electrónica en un 1,3-dipolo consiste en asignar el principal contribuyente de resonancia a partir de datos experimentales o teóricos, tales como mediciones de los momentos dipolares[10] o por cálculos teóricos/computacionales. Por ejemplo, el diazometano tiene por forma resonante más importante (la estructura resonante representativa) la que tiene en el átomo de nitrógeno terminal la carga negativa, mientras que en el ácido hidrazoico la más importante es la que tiene en el átomo de nitrógeno interno el carácter negativo.

Derivados de aziridina editar
Ciertas aziridinas N-sustituidas con grupos aceptores de electrones en ambos carbonos, forman iluros de azometina en una reacción de apertura de anillo térmica o fotoquímica.[11][12] Estos iluros pueden reaccionar con un dipolarófilo adecuado en una cicloadición 1,3-dipolar:[13]

Cuando el grupo N-sustituyente es un grupo aceptor de electrones tal como un grupo tosilo, se rompe el enlace carbono-nitrógeno, formando otro zwitterión: TsN−–CH2–CH2+–R[14]

Con estos reactivos se ha visto como se obtienen derivados de pirrolina.
Óxidos de nitrilo editar
La cicloadición 1,3-dipolar con óxidos de nitrilo es una reacción aldólica enmascarada ampliamente utilizada. La cicloadición entre un óxido de nitrilo y un alqueno da como producto un derivado de isoxazolina, mientras que la reacción con un alquino produce el isoxazol. Ambos productos, isoxazolinas e isoxazoles, pueden ser escindidos por hidrogenación del enlace N-O para revelar la reacción aldólica de tipo β-hidroxicarbonilo o productos β-dicarbonilo de tipo Claisen, respectivamente. La cicloadición del óxido de nitrilo con un alquino, seguido de hidrogenación, se utiliza en la síntesis de Miyakolide como se ilustra en la figura siguiente:[15]

Dipolarófilos editar
Los dipolarófilos más utilizados son los alquenos y alquinos. Los dipolarofilos que contienen heteroátomos, como los carbonilos e iminas, también pueden sufrir cicloadición 1,3-dipolar.
Estereoquímica de la reacción editar
Las reacciones pericíclicas tienen una serie de características, pero la más importante, que es la que caracteriza la estereoquímica de estas reacciones, es que se trata de una reacción concertada en la que el estado de transición tiene una geometría cíclica.
Estereoespecifidad: Retención de configuración editar
Las cicloadiciones 1,3-dipolares suelen dar lugar a la retención de la configuración con respecto tanto al 1,3-dipolo como al dipolarófilo. Este alto grado de estereoespecificidad es lo que apoya el que esta reacción se diga que es concertada (aunque se han descrito mecanismos de reacción por etapas). Como se ha mencionado antes, hay muchos ejemplos que muestran que estas reacciones pueden ser por etapas, por lo tanto, pueden presentar en esos casos una estereoespecificidad parcial o ninguna.
Estereoespecifidad con respecto al dipolarófilo editar
Si nos centramos en el dipolarófilo (concretamente en los alquenos), éste retiene la configuración cis/trans cuando se forma el ciclo como se aprecia en la siguiente figura:[16]

Estereoespecifidad con respecto al 1,3-dipolo editar
En general, la estereoquímica del dipolo no es motivo de gran preocupación porque solo unos pocos dipolos podrían tener centros estereogénicos, y las estructuras de resonancia permiten la rotación de enlaces, lo que rompe la estereoquímica. Sin embargo, el estudio de los iluros de azometina ha verificado que la cicloadición también es estereoespecífica con respecto al componente dipolo. Los iluros de azometina diastereopuros se generan por la apertura del anillo electrocíclico de las aziridinas N-sustituidas, y luego quedan atrapados rápidamente con dipolarófilos fuertes antes de que pueda tener lugar la rotación del enlace (ver esquema a continuación).[17][18] Si se usan dipolarófilos más débiles, los enlaces en el dipolo tienen la posibilidad de rotar, lo que dificulta que exista una estereoespecificidad en la cicloadición con respecto al dipolo.
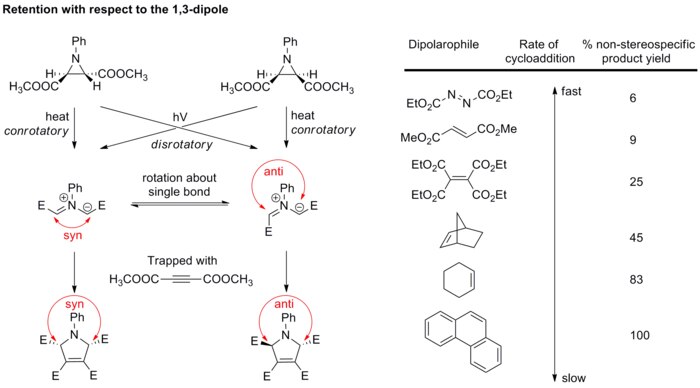
Estos resultados confirman por completo que la cicloadición 1,3-dipolar es estereoespecífica, dando retención tanto del 1,3-dipolo como del dipolarófilo, aunque se deja en evidencia, que esto no es lo normal.
Regioselectividad editar
Por lo general, si el dipolarófilo tiene algún grupo que retira densidad electrónica (grupo electroaceptor), el átomo de carbono unido a él tendrá una densidad electrónica más positiva que la del otro, prefiriendo la unión por el extremo negativo del 1,3-dipolo que por el positivo. Lo mismo ocurre si el dipolarófilo tiene un grupo dador de densidad electrónica, el átomo de carbono unido tendrá una densidad electrónica más negativa que la del otro, de tal manera que preferirá unirse al extremo positivo del 1,3-dipolo. En la realidad, esto se puede hacer con cálculos teóricos, ya que depende de los orbitales moleculares frontera, y no siempre se cumple lo comentado anteriormente.[19] A continuación se pone un ejemplo de esto comentado:

En el ejemplo, se considera la cicloadición de diazometano a tres dipolarófilos: acrilato de metilo, estireno o cinamato de metilo. El carbono del diazometano tiene el orbital HOMO más grande, mientras que los carbonos olefínicos terminales de acrilato de metilo y estireno tienen el mayor LUMO. Así, la cicloadición da la sustitución en la posición C-3 regioselectiva (en la figura los números en rojo son los localizadores del anillo de pirazolina). Para el cinamato de metilo, los dos sustituyentes (Ph vs COOMe) compiten en la retirada de electrones al alqueno. El carboxilo es el mejor grupo de retirada de electrones, causando que el β-carbono sea el más electrofílico. Por lo tanto, cicloadición se obtiene el grupo carboxilo en C-3 y el grupo fenilo en C-4 regioselectiva.
Además, sustituyentes voluminosos tanto en el 1,3-dipolo como en el dipolarófilo pueden producir regioisómeros debido al impedimento estérico que generan estos sustituyentes.
Diastereoselectividad editar
Cuando se generan dos o más centros quirales durante la reacción, se pueden obtener estados de transición y productos diastereoméricos. En la cicloadición de Diels-Alder, generalmente se observa la endo diastereoselectividad debida a interacciones orbitales secundarias. Sin embargo, en las cicloadiciones 1,3-dipolares, dos fuerzas influyen en la diastereoselectividad: la atractiva interacción-π (que se asemeja a las interacciones orbitales secundarias en la cicloadición de Diels-Alder) y la interacción estérica repulsiva. Desafortunadamente, estas dos fuerzas a menudo se cancelan entre sí, causando una mala diastereoselección en la cicloadición 1,3-dipolar.
A continuación se muestran ejemplos de cicloadiciones diastereoselectivas 1,3-dipolares controladas por sustrato. Primero está la reacción entre el N-benciluro de benzonitrilo y el acrilato de metilo. En el estado de transición, los grupos fenilo y éster metílico se apilan para dar la sustitución cis como el producto de pirrolina final exclusivo. Esta interacción-π favorable compensa la repulsión estérica entre los grupos fenilo y éster metílico.[20] La segunda es la reacción entre nitronas y dihidrofurano. La exo-selectividad se logra para minimizar la repulsión estérica.[21] La última es la reacción intramolecular de iluro de azometina con alqueno. La diastereoselectividad se controla mediante la formación de un sistema de anillo fusionado (biciclo) en cis menos tenso.[22]

Referencias editar
- ↑ Milton Orchin, Roger S. Macomber, Allan R. Pinhas, R. Marshall Wilson: The Vocabulary and Concepts of Organic Chemistry. 2. Auflage, John Wiley & Sons, Hoboken 2005, ISBN 0-471-68028-1, S. 583-584.
- ↑ Ivan Ernest: Bindung, Struktur und Reaktionsmechanismen in der organischen Chemie, Springer-Verlag, 1972, S. 349−350, ISBN 3-211-81060-9.
- ↑ Huisgen, Rolf (1963). «1.3-Dipolare Cycloadditionen Rückschau und Ausblick». Angewandte Chemie 785 (13): 604-637. doi:10.1002/ange.19630751304.
- ↑ a b Huisgen, Rolf (noviembre de 1963). «Kinetics and Mechanism of 1,3-Dipolar Cycloadditions». Angewandte Chemie International Edition 2 (11): 633-645. doi:10.1002/anie.196306331. Archivado desde el original el 11 de diciembre de 2012. Consultado el 28 de julio de 2013.
- ↑ Fireston, R (1968). «Mechanism of 1,3-dipolar cycloadditions». Journal of Organic Chemistry 33: 2285-2290. doi:10.1021/jo01270a023.
- ↑ Huisgen, Rolf (1976). «1,3-Dipolar cycloadditions. 76. Concerted nature of 1,3-dipolar cycloadditions and the question of diradical intermediates». Journal of Organic Chemistry 41: 403-419. doi:10.1021/jo00865a001.
- ↑ Huisgen, Rolf (1986). «First Two-Step 1,2-Dipolar Cycloadditons: Nonstereospecificity». J. Am. Chem. Soc. 108: 6401-66402. doi:10.1021/ja00280a053.
- ↑ Blanco-Ania, Daniel (2010). «Synthesis of Dihydrouracils Spiro-Fused to Pyrrolidines: Druglike Molecules Based on the 2-Arylethyl Amine Scaffold». Molecules 15: 2269-2301. doi:10.3390/molecules15042269.
- ↑ Huisgen, Rolf (1963). «1,3-Dipolar Cycloadditions. Past and Future». Angewandte Chemie International Edition 2: 565-598. doi:10.1002/anie.196305651.
- ↑ Sheridan, J (1958). «Microwave Spectra of Diazomethane and its Deutero Derivatives». Nature 181 (4614): 1000-1001. doi:10.1038/1811000a0.
- ↑ Harold W. Heine, Richard Peavy (1965). «Aziridines XI. Reaction of 1,2,3-triphenylaziridine with diethylacetylene dicarboxylate and maleic anhydride». Tetrahedron Letters 6 (35): 3123-6. doi:10.1016/S0040-4039(01)89232-7.
- ↑ Albert Padwa, Lewis Hamilton (1965). «Reactions of aziridines with dimethylacetylene dicarboxylate». Tetrahedron Letters 6 (48): 4363-7. doi:10.1016/S0040-4039(00)71101-4.
- ↑ Philippe Dauban, Guillaume Malik (2009). «A Masked 1,3-Dipole Revealed from Aziridines». Angew. Chem. Int. Ed. 48 (48): 9026-9. doi:10.1002/anie.200904941.
- ↑ Ioana Ungureanua, Cristian Bologab, Saïd Chayera, André Mann (16 de julio de 1999). «Phenylaziridine as a 1,3-dipole. Application to the synthesis of functionalized pyrrolidines». Tetrahedron Letters 40 (29): 5315-8. doi:10.1016/S0040-4039(99)01002-3.
- ↑ Campos, Kevin (1999). «Synthesis and Absolute Stereochemical Assignment of (+)-Miyakolide». Journal of the American Chemical Society 121: 6816-6826. doi:10.1021/ja990789h.
- ↑ Bihlmaier, Werner; Geittner, Jochen; Huisgen, Rolf; ReissigP, Hans-Ulrich (1978). «The Stereospecificity of Diazomethane Cycloadditions». Heterocycles 10: 147-152. doi:10.3987/S-1978-01-0147. Archivado desde el original el 11 de junio de 2019. Consultado el 25 de octubre de 2019.
- ↑ Huisgen, Rolf; Scheer, Wolfgang; Huber, Helmut (1967). «Stereospecific Conversion of cis-trans Isomeric Aziridines to Open-Chain Azomethine Ylides». Journal of the American Chemical Society 89 (7): 1753-1755. doi:10.1021/ja00983a052.
- ↑ Dahmen, Alexander; Hamberger, Helmut; Huisgen, Rolf; Markowski, Volker (1971). «Conrotatory ring opening of cyanostilbene oxides to carbonyl ylides». Journal of the Chemical Society D: Chemical Communications 0 (19): 1192-1194. doi:10.1039/C29710001192.
- ↑ Albert Padwa , Lubor Fisera , Konrad F. Koehler , Augusto Rodriguez , George S. K. Wong (1984). «Regioselectivity associated with the 1,3-dipolar cycloaddition of nitrones with electron-deficient dipolarophiles». J. Org. Chem. 49 (2): 276-281. doi:10.1021/jo00176a012.
- ↑ Padwa, Albert; Smolanoff, Joel (1971). «Photocycloaddition of arylazirenes with electron-deficient olefins». Journal of the American Chemical Society 93 (2): 548-550. doi:10.1021/ja00731a056.
- ↑ Iwashita, Takashi; Kusumi, Takenori; Kakisawa, Hiroshi (1979). «A Synthesis of dl-isoretronecanol». Chemistry Letters 8 (11): 1337-1340. doi:10.1246/cl.1979.1337.
- ↑ Wang, Chia-Lin; Ripka, William; Confalone, Pat (1984). «A short and stereospecific synthesis of (±)-α-lycorane». Tetrahedron Letters 25 (41): 4613-4616. doi:10.1016/S0040-4039(01)91213-4.
 Datos: Q902907
Datos: Q902907 Multimedia: Huisgen cycloaddition / Q902907
Multimedia: Huisgen cycloaddition / Q902907