Timoma
En oncología, el timoma es un tumor que se origina de las células epiteliales del timo y puede ser benigno o maligno. Los timomas son frecuentemente asociados con un desorden neuromúscular llamado miastenia gravis.[1] Una vez diagnosticado, el timoma debe ser resecados quirúrgicamente. En casos de tumor maligno, se puede usar Quimioterapia
| Timoma | ||
|---|---|---|
 Un timoma encapsulado (de tipo linfocítico y epitelial combinado). | ||
| Especialidad | oncología | |
Epidemiología[2] editar
- 0.2-1.5% de todas las neoplasias malignas
- Incidencia general del timoma: 0.15/100.000
- 0.06% de las neoplasias tímicas corresponden a carcinomas
- Mayor prevalencia entre los rangos etarios de 40-60 años
Etiopatogenia editar
El timoma se origina de las células epiteliales que hay en el timo, actualmente se reconocen múltiples variedades histológicas,[1] que dependen de la aparición de estirpes celulares en el microscopio.
- Tipo A (13.2%): si las células epiteliales tienen una forma oval o fusiforme (cuenta linfocitaria baja);
- Tipo B: si tienen una forma epiteloide (el tipo B tiene tres subtipos: B1 (18.4%, rico en linfocitos), B2 (32.9%, cortical) y B3 ( 22.4%, epitelial);[3]
- Tipo AB (7.9%): si el tumor tiene una combinación de ambos tipos
Las células epiteliales corticales del timo tienen un abundante citoplasma, núcleos vesiculares con cromatina finamente dividida y nucleolos pequeños.
Las células epiteliales medulares en contraste, tienen forma de huso con un núcleo oval denso y citoplasma escaso, si recapitula más las características de las células corticales, se cree que es menos benigno.
La enfermedad autoinmunitaria relacionada con el timoma incluye una alteración en los subconjuntos de linfocitos T. La anomalía principal de los linfocitos T parece relacionarse con la adquisición del fenotipo CD45RA+ por los linfocitos T CD4+ indiferenciados durante la timopoiesis intratumoral terminal, seguido de una exportación de estos linfocitos T CD4+ activados hacia la circulación. En el caso de la MG asociada al timoma, se produce una serie de anticuerpos en contra de antígenos neuromusculares, en especial, con el receptor de acetilcolína.[4]
Signos y síntomas editar
Un tercio de todos los pacientes con timoma debutan por síntomas asociados a la compresión de las estructuras que rodean el timo por la masa expansiva. Estos se pueden presentar como un síndrome de la vena cava superior, disfagia, disnea, tos y dolor torácico.[1]
En un tercio de los pacientes este tumor es descubierto debido a que padecen de una enfermedad autoinmune asociada. Como se ha mencionado, la más común de estas enfermedades en la miastenia gravis (MG); del 10-15% de los pacientes con MG tienen un timoma, mientras que 30-45% de pacientes que tienen un timoma padecen de MG.
Otras enfermedades autoinmunes asociadas con este tumor son la autoinmunidad multiorgánica asociada al timoma, la aplasia pura de células rojas, el síndrome de Good (Timoma con inmunodeficiencia combinada e hipogamaglobulinemia). Otras enfermedades asociadas son la pericarditis aguda, agranulocitosis, alopecia areata, colitis ulcerativa, enfermedad de Cushing, anemia hemolítica, encefalopatía límbica, miocarditis, síndrome nefrótico, panhipopituitarismo, anemia perniciosa, polimiositis, artritis reumatoide, sarcoidosis, esclerodermia, radiculopatía sensomotora, lupus eritematoso sistémico y tiroiditis.[1][5]
Del 30 al 50% de las personas con un timoma no tienen síntomas, y la masa es identificada de manera incidental en una radiografía de tórax o en una tomografía computarizada (TC).[1]
Diagnóstico editar

Ante la sospecha de un timoma, generalmente se realiza una TC para valorar el tamaño y extensión del tumor, a este se le puede tomar una muestra a través de una biopsia guiada por radiología. El realce vascular incrementado en la TC puede ser indicativo de malignidad, así como la presencia de derrame pleural.[1] La toma de biopsia está asociada con un pequeño riesgo de neumomediastino o mediastinitis e incluso un riesgo aún menor de daño cardíaco o a los grandes vasos. Algunas veces el timoma puede tener metástasis en el abdomen.[6]
El diagnóstico final es con un estudio histopatológico realizado por un patólogo, después de obtener la muestra del tejido. La clasificación final y el estadio se realiza posterior a la extracción quirúrgica del tumor.
Se pueden utilizar pruebas de laboratorio para buscar problemas asociados o una posible diseminación del tumor. Estos incluyen: hemograma completo, electroforesis de proteínas, anticuerpos contra el receptor de acetilcolina (indicativo de miastenia), electrolitos, enzimas hepáticas y función renal.
Estadio editar
El sistema de estadio de Masaoka es ampliamente usado y se basa en la extensión anatómica de la enfermedad al momento de la cirugía:[7]
- I: completamente encapsulado
- IIA: Invasión microscópica a través de la cápsula hasta el tejido adiposo circundante
- IIB: Invasión macroscópica en la cápsula
- III: Invasión macroscópica en órganos adyacentes
- IVA: Implantes pleurales o pericárdicos
- IVB: Metástasis por vía hematógena o linfática a sitios distantes (extratorácicos)
Tratamiento editar
La extirpación quirúrgica es el estándar de oro para el tratamiento del timoma. Si el tumor es aparentemente invasivo y grande, se sugiere usar quimioterapia y/o radioterapia neoadyuvante (antes de la cirugía) que ayude a disminuir su tamaño y mejorar la resecabilidad. Cuando el tumor esta en estadios tempranos (Masaoka I al IIB), generalmente no es necesaria terapia posterior a la cirugía. En timomas invasivos se sugiere tratamientos adicionales con radioterapia o quimioterapia (ciclofosfamida, doxirrubicina y cisplatino).[1]
La recurrencia del timoma esta descrita hasta en el 10-30% de los casos después de 10 años de la cirugía, y en la mayoría de los casos las recurrencias pleurales pueden ser removidas, posterior a esto se puede usar perfusión de quimioterapia intratorácica hipertérmica.
La resección del timo en los adultos no parece inducir inmunodeficiencia. Sin embargo, en niños, la inmunidad posoperatoria puede ser anormal y se recomienda el uso de varias vacunas contra agentes infecciosos.
Pronóstico editar
El pronóstico es mucho peor en estadios III o IV comparados con los estadios I o II. Los timomas invasivos raramente dan metástasis, generalmente a la pleura, huesos, hígado o cerebro en el 7% de los casos.[1] Un estudio reporta que poco más del 40% de los pacientes en estadio III o IV sobrevivieron al menos 10 años después del diagnóstico. La media de edad al momento del diagnóstico fue de 57 años.[8]
Los pacientes que han tenido una timomectomía deben ser alertados de posibles efectos secundarios después de la vacunación contra la fiebre amarilla. Esto es debido a la respuesta inadecuada de las células T a la vacuna con virus atenuados.
Galería de imágenes editar
-
 An encapsulated cystic thymoma.
An encapsulated cystic thymoma. -
 A locally invasive circumscribed thymoma (mixed lymphocytic and epithelial, mixed polygonal and spindle).
A locally invasive circumscribed thymoma (mixed lymphocytic and epithelial, mixed polygonal and spindle). -
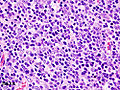 Histopathological image of thymoma type B1. Anterior mediastinal mass surgically resected. Hematoxylin & eosin stain.
Histopathological image of thymoma type B1. Anterior mediastinal mass surgically resected. Hematoxylin & eosin stain. -
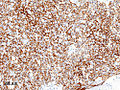 Histopathological image of thymoma type B1. Anterior mediastinal mass surgically resected. Cytokeratin CAM5.2 immunostain.
Histopathological image of thymoma type B1. Anterior mediastinal mass surgically resected. Cytokeratin CAM5.2 immunostain. -
 Histopathological image representing a noninvasive thymoma type B1, surgically resected. Hematoxylin & eosin.
Histopathological image representing a noninvasive thymoma type B1, surgically resected. Hematoxylin & eosin. -
 Thymoma. FNA specimen. Field stain.
Thymoma. FNA specimen. Field stain.


.JPG)
_CK_CAM5-2.JPG)
.JPG)

Referencias editar
- ↑ a b c d e f g h Thomas, Charles R.; Wright, Cameron D.; Loehrer, Patrick J. (1 de julio de 1999). «Thymoma: State of the Art». Journal of Clinical Oncology 17 (7): 2280-2280. ISSN 0732-183X. doi:10.1200/JCO.1999.17.7.2280. Consultado el 30 de noviembre de 2020.
- ↑ Fornasiero A, Daniele O, Ghiotto C, et al.: Chemotherapy of invasive thymoma. J Clin Oncol 8 (8): 1419-23, 1990. [PUBMED Abstract] Engels EA, Pfeiffer RM: Malignant thymoma in the United States: demographic patterns in incidence and associations with subsequent malignancies. Int J Cancer 105 (4): 546-51, 2003.
- ↑ Dadmanesh, F.; Sekihara, T.; Rosai, J. (2001-05). «Histologic typing of thymoma according to the new World Health Organization classification». Chest Surgery Clinics of North America 11 (2): 407-420. ISSN 1052-3359. PMID 11413764. Consultado el 30 de noviembre de 2020.
- ↑ Hoffacker V, Schultz A, Tiesinga JJ, et al.: Thymomas alter the T-cell subset composition in the blood: a potential mechanism for thymoma-associated autoimmune disease. Blood 96 (12): 3872-9, 2000.
- ↑ Bernard, C.; Frih, H.; Pasquet, F.; Kerever, S.; Jamilloux, Y.; Tronc, F.; Guibert, B.; Isaac, S. et al. (1 de enero de 2016). «Thymoma associated with autoimmune diseases: 85 cases and literature review». Autoimmunity Reviews (en inglés) 15 (1): 82-92. ISSN 1568-9972. doi:10.1016/j.autrev.2015.09.005. Consultado el 30 de noviembre de 2020.
- ↑ Geffen, Wouter H. van; Sietsma, Johanna; Roelofs, Pieter MM; Hiltermann, Thijo JN (1 de diciembre de 2011). «A malignant retroperitoneal mass – A rare presentation of recurrent thymoma». Case Reports (en inglés) 2011: bcr0920114737. ISSN 1757-790X. PMC 3229325. PMID 22674945. doi:10.1136/bcr.09.2011.4737. Consultado el 30 de noviembre de 2020.
- ↑ Masaoka, Akira; Monden, Yasumasa; Nakahara, Kazuya; Tanioka, Tsuneo (1981). «Follow-up study of thymomas with special reference to their clinical stages». Cancer (en inglés) 48 (11): 2485-2492. ISSN 1097-0142. doi:10.1002/1097-0142(19811201)48:113.0.CO;2-R. Consultado el 30 de noviembre de 2020.
- ↑ Wilkins, Kirsten Bass; Sheikh, Emran; Green, Rennae; Patel, Mayur; George, Simeon; Takano, Manabu; Diener-West, Marie; Welsh, James et al. (1999-10). «Clinical and Pathologic Predictors of Survival in Patients With Thymoma». Annals of Surgery (en inglés estadounidense) 230 (4): 562. ISSN 0003-4932. PMC 1420905. PMID 10522726. doi:10.1097/00000658-199910000-00012. Consultado el 30 de noviembre de 2020.